
Collaborators: Cintia Fabbri, Marta Giovanetti, Victoria Luppo, Vagner Fonseca, Jorge Garcia, Cintia Barulli, Mariel Feroci, Sofia Perrone, Doraldina Casoni, Sergio Giamperetti, Maria Cristina Alvarez Lopez, Maria Delia Foussal, Mauricio Figueredo, Karina Salvatierra, Sergio Lejona, Natalia Ruiz Diaz, Gonzalo Castro, Gabriela Bravo, Noelia Jackel, Carina Sen, Tomás Poklepovich Caride, Leticia Franco, Carlos Giovachini, Jairo Mendez Rico, Luiz Carlos Junior Alcantara, Maria Alejandra Morales
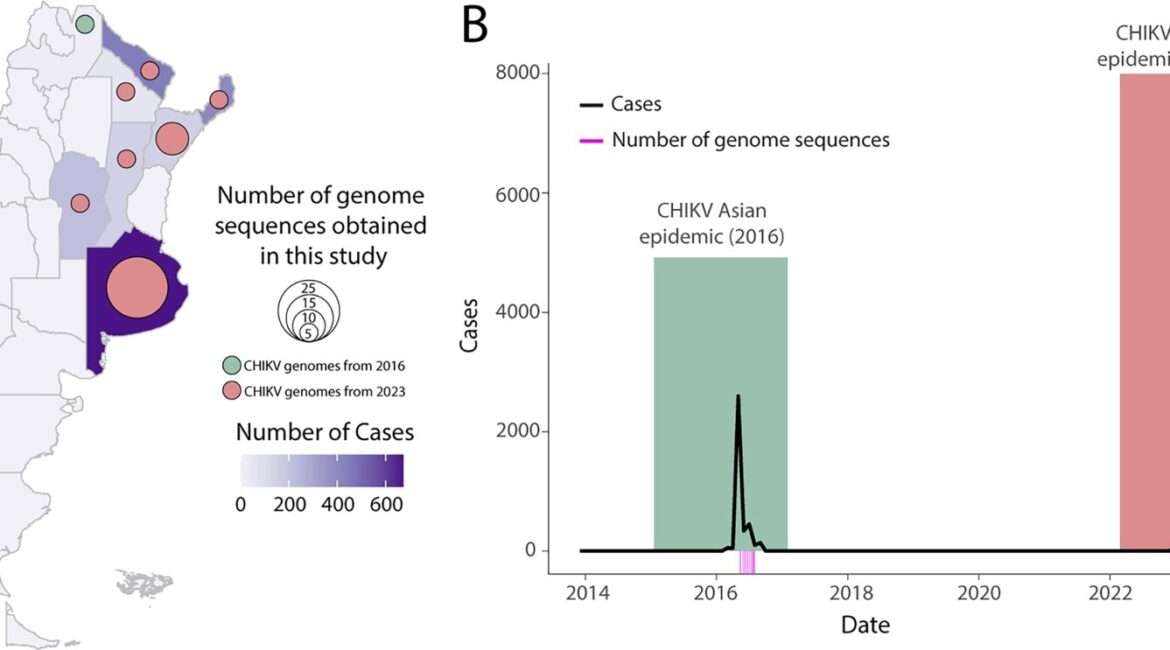
Summary: Chikungunya virus (CHIKV) has emerged as a significant public health concern due to its rapid spread and potential for causing debilitating epidemics. In Argentina, the virus has garnered attention since its introduction to the Americas in 2013, due to its growing incidence and impact in neighbouring countries. Here we present a comprehensive analysis of the spatiotemporal dynamics of CHIKV in Argentina, focusing on the evolutionary trajectory of its genetic variants. Through a combination of active surveillance, screening of historical and recent samples, and whole-genome sequencing, we traced the evolutionary history of CHIKV lineages circulating within the country. Our results reveal that two distinct genotypes circulated in Argentina: The Asian lineage during the 2016 epidemic and the ECSA lineage in 2023. This distribution reflects the dominance of particular variants across Latin America. Since 2023, the ECSA lineage has led to a surge in cases throughout the Americas, marking a significant shift. The replacement of lineages in the American region constitutes a major epidemiological event, potentially affecting the dynamics of virus transmission and the clinical outcomes in impacted populations. The spatiotemporal analysis highlights CHIKV’s distribution across Argentina and underscores the significant role of human mobility, especially when considering recent epidemics in neighbouring countries such as Paraguay and Uruguay, which have facilitated the spread and introduction of the viral strain into different districts. By integrating epidemiological data with genomic insights, we elucidate the patterns of virus dissemination, highlighting key areas of transmission and potential factors contributing to its spread.
Publication Date: 2024-06-10
Journal: Emerging Microbes & Infections
DOI: https://doi.org/10.1080/22221751.2024.2362941